VDJMLpy¶
VDJMLpy is a Python module for working with the results of immune receptor sequence alignment in VDJML format. It is built as bindings to libVDJML, a C++ library.
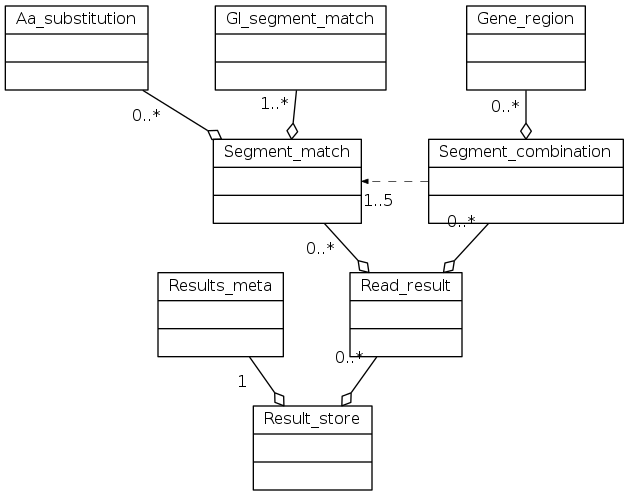
API reference¶
-
class
Aa_substitution¶ amino acid substitution
-
gl_aa((Aa_substitution)arg1) → Aminoacid :¶ amino acid encoded by the germline sequence
-
read_aa((Aa_substitution)arg1) → Aminoacid :¶ amino acid encoded by the read sequence
-
read_position((Aa_substitution)arg1) → int :¶ 0-based read position of the codone’s first nucleotide
-
-
class
Aa_substitutions_set¶ Set of amino acid substitutions
-
empty((Aa_substitutions_set)arg1) → bool :¶ Indicates if there are AA substitutions or not
-
-
class
Aligner_id¶ Aligner software ID
-
class
Aligner_info¶ Info about aligner software
-
id((Aligner_info)arg1) → Aligner_id :¶ Aligner software ID
-
name((Aligner_info)arg1) → str :¶ Aligner software name
-
parameters((Aligner_info)arg1) → str :¶ Parameters used for the alignment
-
run_id((Aligner_info)arg1) → int :¶ Aligner software run id
-
uri((Aligner_info)arg1) → str :¶ Aligner software URI
-
version((Aligner_info)arg1) → str :¶ Aligner software version
-
-
class
Btop¶ Blast trace-back operations, alignment description
-
empty((Btop)arg1) → bool :¶ return True if Btop is empty
-
-
class
Btop_stats¶ Btop_statistics
-
deletions_¶ number of deletions (from read sequence)
-
gl_len_¶ length of the aligned germline sequence
-
insertions_¶ number of insertions (in read sequence)
-
matches((Btop_stats)arg1) → int :¶ number of matches in the alignment
-
read_len_¶ length of the aligned read sequence
-
substitutions_¶ number of substitutions
-
-
class
Codon_match¶ Information about a pair of aligned codons, nucleotide triples, which may potentially contain gaps
-
gl_char((Codon_match)arg1, (int)i) → str :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: codon nucleotide in germline sequence
-
gl_nuc((Codon_match)arg1, (int)i) → Nucleotide :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: codon nucleotide in germline sequence
-
gl_pos((Codon_match)arg1[, (int)i=0]) → int :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: position of codon nucleotide in germline sequence; if a deletion, return position of the first nucleotide to the right, if at end of sequence, return sequence length
-
is_gl_contiguous((Codon_match)arg1) → bool :¶ Returns: True if germline codon contains no gaps
-
is_gl_translatable((Codon_match)arg1) → bool :¶ Returns: True if germline codon can be unambiguously translated
-
is_match((Codon_match)arg1) → bool :¶ Returns: True if same nucleotides in read and germline
-
is_read_contiguous((Codon_match)arg1) → bool :¶ Returns: True if read codon contains no gaps
-
is_read_translatable((Codon_match)arg1) → bool :¶ Returns: True if read codon can be unambiguously translated
-
is_silent((Codon_match)arg1) → bool :¶ Returns: True if is_match()or iftranslate_read()==translate_gl()Raises: RuntimeError – if not is_match()and notis_translatable()
-
is_translatable((Codon_match)arg1) → bool :¶ Returns: True if both codons can be unambiguously translated
-
read_char((Codon_match)arg1, (int)i) → str :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: codon nucleotide in read sequence
-
read_nuc((Codon_match)arg1, (int)i) → Nucleotide :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: codon nucleotide in read sequence
-
read_pos((Codon_match)arg1[, (int)i=0]) → int :¶ Parameters: i (int) – nucleotide position in the codon [0,2] Returns: position of codon nucleotide in read sequence; if a deletion, return position of the first nucleotide to the right, if at end of sequence, return sequence length
-
translate_gl((Codon_match)arg1) → Aminoacid :¶ Returns: amino acid encoded by germline codon Raises: RuntimeError – if not is_gl_translatable()
-
translate_read((Codon_match)arg1) → Aminoacid :¶ Returns: amino acid encoded by read codon Raises: RuntimeError – if not is_read_translatable()
-
-
class
Gene_region¶ information about gene region alignment
-
aligner((Gene_region)arg1) → Aligner_id :¶ aligner ID
-
match_metrics((Gene_region)arg1) → Match_metrics :¶ match metrics
-
numbering_system((Gene_region)arg1) → Numsys_id :¶ numbering system ID
-
read_range((Gene_region)arg1) → Interval :¶ read sequence range
-
region_type((Gene_region)arg1) → Region_id :¶ region ID
-
-
class
Gl_db_id¶ Germline database ID
-
class
Gl_db_info¶ Info about germline sequences database
-
id((Gl_db_info)arg1) → Gl_db_id :¶ germline database ID
-
name((Gl_db_info)arg1) → str :¶ germline database name
-
species((Gl_db_info)arg1) → str :¶ germline database species
-
uri((Gl_db_info)arg1) → str :¶ germline database URI
-
version((Gl_db_info)arg1) → str :¶ germline database version
-
-
class
Gl_seg_id¶ Germline segment ID
-
class
Gl_seg_match_id¶ Germline segment match ID
-
class
Gl_segment_info¶ germline segment description
-
gl_database((Gl_segment_info)arg1) → Gl_db_id :¶ germline database ID
-
id((Gl_segment_info)arg1) → Gl_seg_id :¶ germline segment ID
-
name((Gl_segment_info)arg1) → str :¶ germline segment name
-
segment_type((Gl_segment_info)arg1) → object :¶ germline segment type
-
-
class
Gl_segment_map¶ Map of germline segments aligned to read interval
-
empty((Gl_segment_map)arg1) → bool :¶ Indicates if there are germline segments or not
-
-
class
Gl_segment_match¶ Alignment to germline segment
-
aligner((Gl_segment_match)arg1) → Aligner_id :¶ ID of the software that aligned the germline segment
-
gl_position((Gl_segment_match)arg1) → int :¶ first aligned position index of the germline segment
-
gl_segment((Gl_segment_match)arg1) → Gl_seg_id :¶ germline segment ID
-
id((Gl_segment_match)arg1) → Gl_seg_match_id :¶ germline segment match ID
-
num_system((Gl_segment_match)arg1) → Numsys_id :¶ germline segment numbering system ID
-
-
class
Interval¶ sequence interval
-
static
first_last0((int)first0, (int)last0) → Interval :¶ create interval from 0-based first and last positions
-
static
first_last1((int)first1, (int)last1) → Interval :¶ create interval from 1-based first and last positions
-
last0((Interval)arg1) → int¶
-
last1((Interval)arg1) → int¶
-
length((Interval)arg1) → int¶
-
pos0((Interval)arg1) → int¶
-
static
pos0_len((int)pos0, (int)len) → Interval :¶ create interval from 0-based starting position and length
-
pos1((Interval)arg1) → int¶
-
static
-
class
Match_metrics¶ Metrics of sequence alignment
-
deletions((Match_metrics)arg1) → int :¶ number of bases deleted in the read
-
frame_shift((Match_metrics)arg1) → bool :¶ return True if a frame shift is present (out_frame_indel() || out_frame_vdj())
-
identity((Match_metrics)arg1) → Percent :¶ percent identity
-
insertions((Match_metrics)arg1) → int :¶ number of bases inserted in the read
-
inverted((Match_metrics)arg1) → bool :¶ return True if sequence inverted
-
mutated_invariant((Match_metrics)arg1) → bool :¶ return True if invariant amino acid mutated
-
out_frame_indel((Match_metrics)arg1) → bool :¶ return True if INDEL causes frameshift
-
out_frame_vdj((Match_metrics)arg1) → bool :¶ return True if VDJ rearrangement is out of frame
-
productive((Match_metrics)arg1) → bool :¶ return True if sequence is productive ( ! (out_frame_indel() || out_frame_vdj() || mutated_invariant()) )
-
score((Match_metrics)arg1) → int :¶ alignment score
-
stop_codon((Match_metrics)arg1) → bool :¶ return True if stop codon present
-
substitutions((Match_metrics)arg1) → int :¶ number of base substitutions
-
-
class
Nucleotide_match¶ Information about a pair of aligned nucleotides, which may be a match, substitution, insertion, or deletion
-
gl_nuc((Nucleotide_match)arg1) → str :¶ return nucleotide character in germline sequence; if the mismatch is an insertion, return ‘-‘
-
gl_pos((Nucleotide_match)arg1) → int :¶ return position of nucleotide in germline sequence; if an insertion, return position of the first nucleotide to the right, if at end of sequence, return sequence length
-
is_deletion((Nucleotide_match)arg1) → bool :¶ return True if a deletion (from read sequence)
-
is_insertion((Nucleotide_match)arg1) → bool :¶ return True if insertion (in read sequence)
-
is_match((Nucleotide_match)arg1) → bool :¶ return True if mismatch
-
read_nuc((Nucleotide_match)arg1) → str :¶ return nucleotide character in read sequence; if the mismatch is a deletion, return ‘-‘
-
read_pos((Nucleotide_match)arg1) → int :¶ return position of nucleotide in read sequence; if a deletion, return position of the first nucleotide to the right, if at end of sequence, return sequence length
-
-
class
Numsys_id¶ Numbering system ID
-
class
Read_result¶ Analysis results for one sequencing read
-
id((Read_result)arg1) → str :¶ read ID string
-
insert((Read_result)arg1, (Segment_match)arg2) → Seg_match_id :¶ insert segment match
- insert( (Read_result)arg1, (Segment_combination)arg2) -> None :
- insert combination of segment matches
-
segment_combinations((Read_result)arg1) → Segment_combinations_list :¶ list of segment combinations
-
segment_matches((Read_result)arg1) → Segment_match_map :¶ map of segment matches
-
-
class
Region_id¶ Gene region type ID
-
class
Result_builder¶ Construct alignment results for one sequencing read
-
get((Result_builder)arg1) → Read_result :¶ get result object (internal reference)
-
insert_segment_combination((Result_builder)arg1, (Seg_match_id)seg_match_1[, (Seg_match_id)seg_match_2=<vdjml._vdjml_py.Seg_match_id object at 0x7f84d76c5de0>[, (Seg_match_id)seg_match_3=<vdjml._vdjml_py.Seg_match_id object at 0x7f84d76c5d70>[, (Seg_match_id)seg_match_4=<vdjml._vdjml_py.Seg_match_id object at 0x7f84d76c5d00>[, (Seg_match_id)seg_match_5=<vdjml._vdjml_py.Seg_match_id object at 0x7f84d76c5c90>]]]]) → Segment_combination_builder¶
-
insert_segment_match((Result_builder)arg1, (int)read_pos0, (str)btop, (str)vdj, (str)seg_name, (int)gl_pos0[, (Match_metrics)metric=<vdjml._vdjml_py.Match_metrics object at 0x7f84db3e8938>[, (Gl_db_id)gl_database=<vdjml._vdjml_py.Gl_db_id object at 0x7f84d76c5f30>[, (Numsys_id)num_system=<vdjml._vdjml_py.Numsys_id object at 0x7f84d76c5ec0>[, (Aligner_id)aligner=<vdjml._vdjml_py.Aligner_id object at 0x7f84d76c5e50>]]]]) → Segment_match_builder :¶ add new segment match
-
release((Result_builder)arg1) → Read_result :¶ get final result object (independent copy); Result_builder object cannot be used anymore
-
-
class
Result_factory¶ Construct alignment results for many sequencing reads
-
new_result((Result_factory)arg1, (str)read_id) → Result_builder :¶ new result builder
-
set_default_aligner((Result_factory)arg1, (Aligner_id)arg2) → None :¶ set default aligner
- set_default_aligner( (Result_factory)arg1, (str)name, (str)version [, (str)parameters=’’ [, (str)uri=’’ [, (int)run_id=0]]]) -> Aligner_id :
- set default aligner
-
set_default_gl_database((Result_factory)arg1, (Gl_db_id)arg2) → None :¶ set default germline database
- set_default_gl_database( (Result_factory)arg1, (str)name, (str)version, (str)species [, (str)url=’‘]) -> Gl_db_id :
- set default germline database
-
set_default_num_system((Result_factory)arg1, (Numsys_id)arg2) → None :¶ set default numbering system
- set_default_num_system( (Result_factory)arg1, (str)name) -> Numsys_id :
- set default numbering system
-
-
class
Result_store¶ Storage of sequencing read results
-
empty((Result_store)arg1) → bool¶
-
insert((Result_store)arg1, (Read_result)arg2) → None :¶ add new result
-
meta((Result_store)arg1) → Results_meta¶
-
-
class
Results_meta¶ Metadata for a collection of alignment results of sequencing reads
-
aligner_map((Results_meta)arg1) → Aligner_map :¶ return a map of aligner software descriptions
-
gene_region_map((Results_meta)arg1) → Gene_region_map :¶ return a map of gene region descriptions
-
gl_db_map((Results_meta)arg1) → Germline_db_map :¶ return a map of germline database descriptions
-
gl_segment_map((Results_meta)arg1) → Gl_segment_map :¶ return a map of germline segment descriptions
-
insert((Results_meta)arg1, (Aligner_info)arg2) → Aligner_id :¶ insert information about aligner software
- insert( (Results_meta)arg1, (Gl_db_info)arg2) -> Gl_db_id :
- insert information about database of germline segments
- insert( (Results_meta)arg1, (Gl_segment_info)arg2) -> Gl_seg_id :
- insert information about germline segment
-
num_system_map((Results_meta)arg1) → Num_system_map :¶ return a map of numbering systems
-
-
class
Seg_match_id¶ Segment match ID
-
class
Segment_combination¶ combination of aligned germline segments
-
insert((Segment_combination)arg1, (Seg_match_id)arg2) → None :¶ insert segment match ID
- insert( (Segment_combination)arg1, (Gene_region)arg2) -> None :
- insert gene region
-
regions((Segment_combination)arg1) → Gene_region_set :¶ collection of gene regions
-
segments((Segment_combination)arg1) → Segment_match_id_set :¶ set of segment match IDs
-
-
class
Segment_combination_builder¶ Construct alignment results for a combination of germline gene segments
-
insert_region((Segment_combination_builder)arg1, (str)name, (Interval)read_range[, (Match_metrics)metric=<vdjml._vdjml_py.Match_metrics object at 0x7f84db3e88c0>[, (Numsys_id)num_system=<vdjml._vdjml_py.Numsys_id object at 0x7f84d76c5980>[, (Aligner_id)aligner_id=<vdjml._vdjml_py.Aligner_id object at 0x7f84d76c5910>]]]) → None¶ - insert_region( (Segment_combination_builder)arg1, (Region_id)region, (Interval)read_range [, (Match_metrics)metric=<vdjml._vdjml_py.Match_metrics object at 0x7f84db3e8848> [, (Numsys_id)num_system=<vdjml._vdjml_py.Numsys_id object at 0x7f84d76c58a0> [, (Aligner_id)aligner_id=<vdjml._vdjml_py.Aligner_id object at 0x7f84d76c5830>]]]) -> None :
indicate gene region location in read sequence
param region: Region_id, type of regionparam read_range: Interval, start and end positions in read sequenceparam metric: Match_metrics, alignment metrics between read and germline sequences; default: no metrics recordedparam num_system: Numsys_id, default: no numbering system recordedparam aligner_id: Aligner_id, default: current aligner ID is used
-
-
class
Segment_match¶ Alignment results for a read segment
-
aa_substitutions((Segment_match)arg1) → Aa_substitutions_set :¶ amino acid substitutions
-
btop((Segment_match)arg1) → Btop :¶ BTOP alignment description
-
gl_length((Segment_match)arg1) → int :¶ length of the aligned germline segment(s)
-
gl_range((Segment_match)arg1) → Interval :¶ nucleotide range of the first germline sequence that matches the read sequence
- gl_range( (Segment_match)arg1, (Gl_segment_match)arg2) -> Interval :
- nucleotide range of the specified germline sequence that matches the read sequence
-
gl_segments((Segment_match)arg1) → Gl_segment_map :¶ germline segment map
-
id((Segment_match)arg1) → Seg_match_id :¶ segment match ID
-
insert((Segment_match)arg1, (Gl_segment_match)arg2) → Gl_seg_match_id :¶ insert germline segment match
- insert( (Segment_match)arg1, (Aa_substitution)arg2) -> None :
- insert amino acid substitution
-
match_metrics((Segment_match)arg1) → Match_metrics :¶ alignment metrics
-
read_range((Segment_match)arg1) → Interval :¶ read sequence nucleotide range that matches to germline segment
-
-
class
Segment_match_builder¶ Construct alignment results for one sequencing read segment match
-
get((Segment_match_builder)arg1) → Segment_match :¶ get segment match structure
-
insert_aa_substitution((Segment_match_builder)arg1, (int)read_pos0, (str)read_aa, (str)gl_aa) → None :¶ add amino acid substitution information
- insert_aa_substitution( (Segment_match_builder)arg1, (int)read_pos0, (str)read_aa, (str)gl_aa) -> None :
- add amino acid substitution information
-
insert_gl_segment_match((Segment_match_builder)arg1, (Gl_seg_id)gl_segment_id, (int)pos0[, (Numsys_id)num_system_id=<vdjml._vdjml_py.Numsys_id object at 0x7f84d76c5c20>[, (Aligner_id)aligner=<vdjml._vdjml_py.Aligner_id object at 0x7f84d76c5bb0>]]) → Gl_seg_match_id :¶ add germline segment alignment info
- insert_gl_segment_match( (Segment_match_builder)arg1, (str)vdj, (str)seg_name, (int)gl_pos0 [, (Gl_db_id)gl_database=<vdjml._vdjml_py.Gl_db_id object at 0x7f84d76c5b40> [, (Numsys_id)num_system=<vdjml._vdjml_py.Numsys_id object at 0x7f84d76c5ad0> [, (Aligner_id)aligner=<vdjml._vdjml_py.Aligner_id object at 0x7f84d76c5a60>]]]) -> Gl_seg_match_id :
- add germline segment alignment info
-
-
class
Sequence_match¶ Aligned sequences, start and end indices
-
end_¶ last plus one position for aligned read and germline sequences
-
seq_¶ aligned read and germline sequences
-
start_¶ 0-based starting position for aligned read and germline sequences
-
-
class
Vdjml_generator_info¶ Info about aligner software
-
datetime((Vdjml_generator_info)arg1) → object :¶ file creation date and time, GMT
-
datetime_str((Vdjml_generator_info)arg1) → str :¶ file creation date and time, GMT
-
name((Vdjml_generator_info)arg1) → str :¶ VDJML file generator name
-
version((Vdjml_generator_info)arg1) → str :¶ VDJML file generator version
-
-
class
Vdjml_reader¶ Incrementally parse VDJML read-by-read
-
generator_info((Vdjml_reader)arg1) → Vdjml_generator_info :¶ information about VDJML generator
-
has_result((Vdjml_reader)arg1) → bool :¶ return True if result was found
-
meta((Vdjml_reader)arg1) → Results_meta :¶ results meta
-
next((Vdjml_reader)arg1) → None :¶ parse next read result
-
result((Vdjml_reader)arg1) → Read_result :¶ return parsed result
-
version((Vdjml_reader)arg1) → int :¶ VDJML version of the file
-
version_str((Vdjml_reader)arg1) → str :¶ VDJML version of the file
-
-
class
Vdjml_writer¶ Incrementally serialize VDJ alignment results
-
class
Xml_writer_options¶ Options for XML output
-
buff_size¶ output buffer size
-
indent¶ indentation string
-
quote¶ quotation character
-
-
codons((Btop)btop[, (int)read_start=18446744073709551615L[, (int)gl_start=18446744073709551615L[, (str)read_seq=''[, (str)gl_seq=''[, (bool)follow_read=False[, (bool)follow_gl=False[, (str)match_char='.']]]]]]]) → object :¶ Returns: codon iterator
-
mismatches((Btop)btop) → Mismatch_iter :¶ return nucleotide mismatch iterator
-
nucleotide_match((Btop)btop[, (int)read_pos0=18446744073709551615L[, (int)gl_pos0=18446744073709551615L[, (str)read_seq=''[, (str)gl_seq=''[, (str)match_char='.']]]]]) → Nucleotide_match :¶ Provides information about a pair of aligned nucleotides
param btop: BtopBTOP structureparam read_pos0: 0-based position relative to read sequence param gl_pos0: 0-based position relative to germline sequence param read_seq: read sequence param gl_seq: germline sequence param match_char: char, character to indicate matching nucleotidesreturn: Nucleotide_matchinformation about two aligned nucleotides- nucleotide_match( (Segment_match)sm, (object)pos0 [, (Gl_segment_match)gsm]) -> Nucleotide_match :
- Generate
Nucleotide_match, information about two aligned
-
nucleotides((Btop)btop[, (str)read_seq=''[, (str)gl_seq=''[, (str)match_char='.']]]) → object :¶ return nucleotide iterator
-
numbering_system((Gl_segment_match)gl_segment_match, (Results_meta)meta) → str :¶ return numbering system name
-
segment_name((Gl_segment_match)gl_segment_match, (Results_meta)meta) → str :¶ return segment name
-
segment_type((Gl_segment_match)gl_segment_match, (Results_meta)meta) → str :¶ return numbering system name
-
sequence_match((Btop)btop[, (int)read_start=18446744073709551615L[, (int)read_end=18446744073709551615L[, (int)gl_start=18446744073709551615L[, (int)gl_end=18446744073709551615L[, (str)read_seq=''[, (str)gl_seq=''[, (str)match_char='.']]]]]]]) → Sequence_match :¶ Generate a pair of aligned sequences with positions for start and end
Parameters: - btop –
Btop, BTOP structure - read_start – position for alignment start (0-based, relative to read sequence)
- read_end – position for alignment end (0-based, relative to read sequence)
- gl_start – position for alignment start (0-based, relative to germline sequence)
- gl_end – position for alignment end (0-based, relative to germline sequence)
- read_seq – read sequence
- gl_seq – germline sequence
- match_char –
char, character to indicate matching nucleotides
Returns: - btop –
-
trim_complement((str)seq, (Interval)interval, (bool)reverse) → str :¶ Trim and optionally reverse-complement a sequence
-
write_to_file((str)path, (Result_store)store[, (Compression)compression=vdjml._vdjml_py.Compression.Unknown_compression[, (int)version=1000[, (Xml_writer_options)options=<vdjml._vdjml_py.Xml_writer_options object at 0x7f84db3de2d8>]]]) → None¶
vdjml/python/igblast_parse.py is part of VDJML project Distributed under the Boost Software License, Version 1.0; see doc/license.txt. Copyright, The University of Texas Southwestern Medical Center, 2014 Author Edward A. Salinas 2014
-
comp_dna(dna, allowIUPAC=False)¶ complement a string (of DNA) allow IUPAC complementing if desired
-
compareIGBlastJuncDataWithQueryJuncData(query_rec, igblastSeq, igBlastInterval, inverted_flag)¶ The basic idea of this suburoutine is as follows: 1) Assuming the query read is given proceed to step #2 2) Recive the junction interval passed in (computed by the code) AND receive the junction sequence passed in (given by IGBLAST) 3) compare the sequence given by IgBLAST with the sequence extracted from the read (using the computed interval taking into account inversion or not) 4) if the SEQUENCE extracted from the read from the computed interval does NOT match the sequence as given by IgBLAST, then print out an error message
-
extractAsItemOrFirstFromList(t)¶ If an item is a list, return the first item if it’s found if the item is not a list, just return the item
-
extractJunctionRegionSeq(jRegion, query_rec)¶ given a pyVDJML junction region, return the sequece as it would appear in IGBLAST output
-
extractSubSeq(interval, seq, is_inverted)¶ given a seq record, an interval into it, and an inverted flag (telling whether interval is in the opposite strand or not) return the subsequence (inclusive) indicated by the interval NOTE that the interval has 1-based indices
-
getRevCompInterval(i, seq_len_in_bp)¶ given an interval (in list form [from,to]) with 1-based indesing AND given a sequence length in BP return the same interval but on the reverse strand
form an interval in one strand find the interval in the reverse complement strand
-
getSubstitutionsInsertionsDeletionsFromBTOP(btop)¶ from a BTOP string extract the numbers of insertions, deletions, and substitutions in the BTOP for the pairs, the first character belongs to the READ(query), the second to the GERMLINE(subject)
-
makeMap(col_list, val_tab_str)¶ from a column list (keys) and tab-separated values make a dict/map
-
makeMetricFromCharMap(charMap)¶ given a characterization map for a region, make a metrics object
-
obtainJuncIntervalAndSeq(juncMapKey, juncMap, juncFirstStartSegMap, juncFirstEndSegMap, isVJJunc=False)¶ Analyze the junction sequence and look at the areas surrounding the junction (anchors on each end VJ, DJ, VD) Based on the analysis compute a read interval and declare it to be the junction. Return that interval as well as the sequence as a package via an array. Testing of this code with real data (millions of reads) has shown that when intervals and sequences are returned that when the interval is used with the actual read that the returned sequences from here match the sequences retrieved from the read using the computed interval.
-
printMap(m)¶ little utility to print a map
-
scanOutputToVDJML(input_file, fact, fasta_query_path=None)¶ Scan lines of IgBLAST output Use # at the beginning of lines to identify IgBLAST sections of output. Based on those sections interpret/classify the output (as alignment summary or hit data for example) and package/accumulate the data. Then, once the end of a record is reached (as indicated by processing the number of hits as it said it got) Send the accumulated/aggreagated/packaged data to vdjml_read_serialize to turn the data into a PyVDJML object. And then return that created/serialized object